For this, we determined if DMOG can protect MEFs lacking both the Suv39h1 and Suv39h2 methyltransferases. The ability of DMOG and CoCl2 to increase Hif1a levels was not altered in the SuvDKO MEFs. Further, whereas DMOG functioned as a radioprotector in normal MEFs, the protective effect of DMOG was significantly reduced in the SuvDKO MEFs. This indicates that the reduced ability of DMOG to protect SuvDKO MEFs may be attributed to both the loss of Hif1a dependent increase in the Suv39h1 methyltransferase, as well as changes in other Hif1a target genes. Overall, figure 5 demonstrates that DMOG can protect cells form radiation by stabilizing the Hif1a transcription factor, and increasing expression of genes, including the Suv39h1 methyltransferase, which can promote cell survival. Finally, we sought to determine if DMOG could protect cells from DNA damage at the level of the whole organism. Hif1a activation can transcriptionally activate a wide range of genes, including VEGF, which promote angiogenesis, and erythropoietin, which mobilizes bone marrow and improves blood parameters. It is likely that these factors may be important in promoting recovery of sensitive tissues, such as the bone marrow and gastrointestinal tract, from total body irradiation. In order to assess if the radioprotective effects of DMOG can be detected at the level of the whole organism, we examined if DMOG had protective effects in a murine total body irradiationmodel. DMOG was injected ip into either C57BL/6J or Balb/c mice, two widely used murine TBI model systems. DMOG alone did not cause any toxicity, with 100% of the mice surviving at 30 days, in agreement with previous studies using DMOG. TBI at 8Gy caused 80% and 100% lethality in saline-treated C57BL/6J and Balb/c mice respectively. However, DMOG treatment significantly improved the survival of both C57BL/6J and Balb/c mice and rescued them ![]() from radiation induced lethality. Activation of Hif1a by DMOG results in the transcriptional PR-171 upregulation of many genes and growth factors and is associated with increased cell survival in culture and protection of mice from whole body irradiation. These results clearly demonstrated that DMOG exhibits radioprotective potential in murine models. The Hif1a transcriptional regulatory protein controls the expression of genes which promote cell survival under conditions of low oxygen tension. Hif1a can switch cell metabolism towards increased expression of growth factors and anti-apoptotic factors as well as switching of metabolic pathways, allow the cell to maintain energy levels under hypoxic conditions. Hif1a levels are controlled through direct hydroxylation of Hif1a by the PHD2 prolylhydroxylase, leading to degradation of Hif1a under normal oxygen tension. Small molecule inhibitors of prolylhydroxylases have been developed which inhibit PHD2 and related enzymes, leading to stabilization of Hif1a and upregulation of the hypoxia response. Here, we have shown that stabilization of Hif1a using the prolylhydroxylase inhibitor DMOG protects both MEFs and mice from the cytotoxic effects of exposure to IR. This is consistent with previous studies high throughput screening demonstrating that tumor cells containing constitutively high levels of Hif1a are more resistant to both chemotherapy and radiotherapy. Increasing Hif1a levels in normal cells with prolylhydroxylase inhibitors such as DMOG therefore represents a novel pathway for the development of new and effective radioprotective agents. Our results clearly show that DMOG requires Hif1a to protect cells from radiation, since suppression of Hif1a with shRNA abolished the protective effect of DMOG.
from radiation induced lethality. Activation of Hif1a by DMOG results in the transcriptional PR-171 upregulation of many genes and growth factors and is associated with increased cell survival in culture and protection of mice from whole body irradiation. These results clearly demonstrated that DMOG exhibits radioprotective potential in murine models. The Hif1a transcriptional regulatory protein controls the expression of genes which promote cell survival under conditions of low oxygen tension. Hif1a can switch cell metabolism towards increased expression of growth factors and anti-apoptotic factors as well as switching of metabolic pathways, allow the cell to maintain energy levels under hypoxic conditions. Hif1a levels are controlled through direct hydroxylation of Hif1a by the PHD2 prolylhydroxylase, leading to degradation of Hif1a under normal oxygen tension. Small molecule inhibitors of prolylhydroxylases have been developed which inhibit PHD2 and related enzymes, leading to stabilization of Hif1a and upregulation of the hypoxia response. Here, we have shown that stabilization of Hif1a using the prolylhydroxylase inhibitor DMOG protects both MEFs and mice from the cytotoxic effects of exposure to IR. This is consistent with previous studies high throughput screening demonstrating that tumor cells containing constitutively high levels of Hif1a are more resistant to both chemotherapy and radiotherapy. Increasing Hif1a levels in normal cells with prolylhydroxylase inhibitors such as DMOG therefore represents a novel pathway for the development of new and effective radioprotective agents. Our results clearly show that DMOG requires Hif1a to protect cells from radiation, since suppression of Hif1a with shRNA abolished the protective effect of DMOG.
The Hif1a dependent increase in expression of the Suv39h1 methyltransferase was required for DMOG dependent radioprotection
Leave a reply
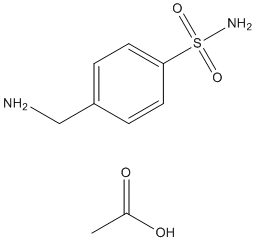 redistribution approaches, as cell-based assay technology that uses protein translocation as the primary readout have been used to study the activity of cellular signaling pathways. Protein targets are labeled with autofluorescent proteins and are read using high-throughput, microscope-based instruments. Although, protein translocation assays have the potential for high-content, high-throughput screeningapplications, such assays are generally not used for proteases. Here, the spatial and functional division into the nucleus and the cytoplasm was exploited to design a translocation-based Taspase1-biosensor assay. The CBA was adapted on a HTS platform, employed to identify potential Taspase1 small molecule inhibitors, and was used to study Taspase1 function in living cells. The robust performance of the TS-Cl2+CBA met critical requirements for high content screening: the biosensor was nontoxic, localized to the cytoplasm in the absence of ectopically expressed Taspase1, and efficiently accumulated in the nucleus following Taspase1-specific cleavage. Hence, we tested whether the assay can also be used on a high-throughput microscopy based screening platform. As cell lines inducibly expressing biosensors may facilitate certain HCS/HTS applications, we generated stable Tet-off TSCl2 + TRE cell lines. The tetracycline -regulated system has been used successfully in various applications. Whereas expression of TS-Cl2 + TRE was blocked in the presence of doxycycline, Dox removal induced TS-Cl2+ TRE expression. Cleavage of TS-Cl2+TRE by the endogenous Taspase1 subsequently resulted in nuclear accumulation of the biosensor.
redistribution approaches, as cell-based assay technology that uses protein translocation as the primary readout have been used to study the activity of cellular signaling pathways. Protein targets are labeled with autofluorescent proteins and are read using high-throughput, microscope-based instruments. Although, protein translocation assays have the potential for high-content, high-throughput screeningapplications, such assays are generally not used for proteases. Here, the spatial and functional division into the nucleus and the cytoplasm was exploited to design a translocation-based Taspase1-biosensor assay. The CBA was adapted on a HTS platform, employed to identify potential Taspase1 small molecule inhibitors, and was used to study Taspase1 function in living cells. The robust performance of the TS-Cl2+CBA met critical requirements for high content screening: the biosensor was nontoxic, localized to the cytoplasm in the absence of ectopically expressed Taspase1, and efficiently accumulated in the nucleus following Taspase1-specific cleavage. Hence, we tested whether the assay can also be used on a high-throughput microscopy based screening platform. As cell lines inducibly expressing biosensors may facilitate certain HCS/HTS applications, we generated stable Tet-off TSCl2 + TRE cell lines. The tetracycline -regulated system has been used successfully in various applications. Whereas expression of TS-Cl2 + TRE was blocked in the presence of doxycycline, Dox removal induced TS-Cl2+ TRE expression. Cleavage of TS-Cl2+TRE by the endogenous Taspase1 subsequently resulted in nuclear accumulation of the biosensor.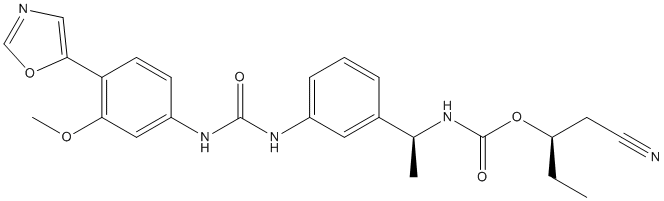 death at an MOI of 0.01in a dosedependent manner. Moreover, maintenance of MDCK cells in the presence of iota-carrageenan up to 96 hours post infection with H1N1 also resulted in a dramatic reduction of viral titers by 2-4 logs, indicative of a protective effect of iota-carrageenan with regard to the spread and release of viral particles from previously infected MDCK cells. However, an increased amount of input virus gradually reduces the protective effect. Therefore, we conclude that the antiviral effect of carrageenan is dependent on the relative amount of input virus in both cases.
death at an MOI of 0.01in a dosedependent manner. Moreover, maintenance of MDCK cells in the presence of iota-carrageenan up to 96 hours post infection with H1N1 also resulted in a dramatic reduction of viral titers by 2-4 logs, indicative of a protective effect of iota-carrageenan with regard to the spread and release of viral particles from previously infected MDCK cells. However, an increased amount of input virus gradually reduces the protective effect. Therefore, we conclude that the antiviral effect of carrageenan is dependent on the relative amount of input virus in both cases.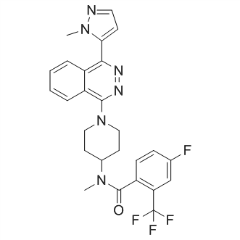 C-terminal interdomain cleft formed by residues from the C-terminal b-sheet, T7-loop and H7-helix, offers an alternative opportunity for the design of FtsZ inhibitors with therapeutic potential in antibiotic-resistant bacterial diseases. Berberine is a plant alkaloid with a long history of medicinal use in traditional Chinese and native American medicines. Berberine extracts show significant antimicrobial activity against bacteria, viruses and fungi. Its potential mechanisms of antimicrobial activity include the suppression of cell adhesion and migration, and inhibition of microbial enzymes. Moreover, recent literature reports demonstrated that berberine is active against Gram-positive bacteria with minimum inhibitory concentration values in the range of 100�C 400 mg/mL by targeting the cell division protein FtsZ. Therefore, berberine is an attractive lead for the development of potent FtsZ inhibitors. Given the availability of X-ray crystal structures of FtsZ, molecular docking is particularly appealing for guiding the chemical derivatization of berberine. Previous studies suggested that berberine binds FtsZ in a hydrophobic pocket. In this paper we report the design and biological study of a series of 9-phenoxyalkyl berberine derivatives with potent inhibition of FtsZ GTPase activity and broadspectrum of antibacterial activity. Natural products and semi-synthetic derivatives provide a rich source of bioactive compounds for the development of new antibacterial agents. However, in the past decade most of the new chemical entities that reached the clinical practice were derived from the same natural scaffolds. Berberine has been traditionally used to treat microbial infections. At the doses commonly used, the compound is considered safe.
C-terminal interdomain cleft formed by residues from the C-terminal b-sheet, T7-loop and H7-helix, offers an alternative opportunity for the design of FtsZ inhibitors with therapeutic potential in antibiotic-resistant bacterial diseases. Berberine is a plant alkaloid with a long history of medicinal use in traditional Chinese and native American medicines. Berberine extracts show significant antimicrobial activity against bacteria, viruses and fungi. Its potential mechanisms of antimicrobial activity include the suppression of cell adhesion and migration, and inhibition of microbial enzymes. Moreover, recent literature reports demonstrated that berberine is active against Gram-positive bacteria with minimum inhibitory concentration values in the range of 100�C 400 mg/mL by targeting the cell division protein FtsZ. Therefore, berberine is an attractive lead for the development of potent FtsZ inhibitors. Given the availability of X-ray crystal structures of FtsZ, molecular docking is particularly appealing for guiding the chemical derivatization of berberine. Previous studies suggested that berberine binds FtsZ in a hydrophobic pocket. In this paper we report the design and biological study of a series of 9-phenoxyalkyl berberine derivatives with potent inhibition of FtsZ GTPase activity and broadspectrum of antibacterial activity. Natural products and semi-synthetic derivatives provide a rich source of bioactive compounds for the development of new antibacterial agents. However, in the past decade most of the new chemical entities that reached the clinical practice were derived from the same natural scaffolds. Berberine has been traditionally used to treat microbial infections. At the doses commonly used, the compound is considered safe.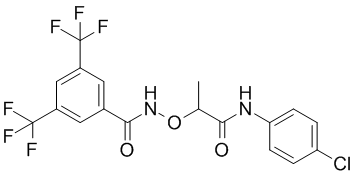 an elastic modulus of approximately 1 GPa, whereas tissues in the body are less than 100 kPa, with brain having an elasticity less than 1 kPa, muscle around 10 kPa, and bone around 100 kPa. The effects of matrix stiffness are typically evaluated by analyzing cell behavior in different gel systems. Stiffness or elasticity can be varied by simply changing the crosslinking
an elastic modulus of approximately 1 GPa, whereas tissues in the body are less than 100 kPa, with brain having an elasticity less than 1 kPa, muscle around 10 kPa, and bone around 100 kPa. The effects of matrix stiffness are typically evaluated by analyzing cell behavior in different gel systems. Stiffness or elasticity can be varied by simply changing the crosslinking