Likely, cell-cell adhesion corrects, to some degree, the imbalanced Rac and Rho activities in Cinoxacin Du145KAI1/CD82 cells as cell-cell contacts typically promote Rac activity and cortical actin network formation but suppress Rho activity and stress fiber formation. Hence, KAI1/CD82 likely induces the motility-related morphological and cytoskeletal phenotypes 1) only when cell-cell adhesion falls apart and 2) through cell-ECM adhesion. In other words, KAI1/CD82 probably inhibits only the 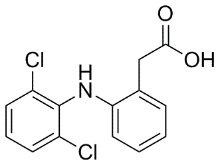 movement of isolated individual cells. In conclusion, we demonstrated in this study that KAI1/CD82 likely intercepts multiple signaling pathways at the plasma membrane by perturbing membrane lipids. This perturbation results in the imbalance of Rac, Rho, and their effector activities and the aberrancy in actin organization and reorganization. The cytoskeletal abnormality in KAI1/CD82-expressing cancer cells causes the attenuation of both cellular protrusion and retraction processes, which ultimately leads to the suppression of cell movement. Although Butenafine hydrochloride tetraspanins physically associate with each other and functionally crosstalk, each tetraspanin has specific functional identity, evidenced by the unique phenotypes displayed by the knockout mouse lines of several tetraspanins. Also, our earlier study underpinned that CD82-mediated inhibition of cell movement is independent of other tetraspanins. Thus, the motility-inhibitory mechanism described above appears to be specific to KAI1/CD82. High-throughput RNA-seq using next-generation Illumina sequencing technique provides an effective and comprehensive method to deeply study the transcriptome of an organism such as human, yeast, rice, oriental fruit fly and Tetrahymena thermophila. With the development of sequencing technique and mycology, the genomes or transcriptomes of fungi have been increasingly obtained for further research, including Metarhizium species, Coprinus cinerea, Trichoderma reesei, Schizophyllum commune, Laccaria bicolor, Tuber melanosporum, Postis placenta. By mapping the sequenced transcriptome reads to a reference genome, the structure annotation can be improved and novel transcripts and alternative splicing can be predicted, which expand the current genome databases. Proteomic one-dimensional or two-dimensional gel electrophoresis approaches combined with mass spectrometry have been widely used to identify interested proteins in fungi and many other organisms. Since the expression of some genes is regulated at the transcriptional level while some other genes at the translational level, the combination of transcriptome and proteome analysis is required and has been widely applied. In this study, we conducted the transcriptome and proteome analysis of different developmental stages of C. militaris and fruiting body cultured on rice medium). About 68 million reads were obtained from each sample and were mapped to the C. militaris genome. We found 2712 differentially expressed genes between mycelium and fruiting body. Functional annotations were used to investigate their difference including the cordycepin metabolism difference. In addition, 359 and 214 non-redundant proteins were identified from mycelium and fruiting body respectively by nESI-LC-MS/ MS. Proteome analysis revealed the developmental-associated protein expression patterns including some noteworthy proteins. Our transcriptome and proteome analysis data enrich the current database of C. militaris and will facilitate the future developmental study.
movement of isolated individual cells. In conclusion, we demonstrated in this study that KAI1/CD82 likely intercepts multiple signaling pathways at the plasma membrane by perturbing membrane lipids. This perturbation results in the imbalance of Rac, Rho, and their effector activities and the aberrancy in actin organization and reorganization. The cytoskeletal abnormality in KAI1/CD82-expressing cancer cells causes the attenuation of both cellular protrusion and retraction processes, which ultimately leads to the suppression of cell movement. Although Butenafine hydrochloride tetraspanins physically associate with each other and functionally crosstalk, each tetraspanin has specific functional identity, evidenced by the unique phenotypes displayed by the knockout mouse lines of several tetraspanins. Also, our earlier study underpinned that CD82-mediated inhibition of cell movement is independent of other tetraspanins. Thus, the motility-inhibitory mechanism described above appears to be specific to KAI1/CD82. High-throughput RNA-seq using next-generation Illumina sequencing technique provides an effective and comprehensive method to deeply study the transcriptome of an organism such as human, yeast, rice, oriental fruit fly and Tetrahymena thermophila. With the development of sequencing technique and mycology, the genomes or transcriptomes of fungi have been increasingly obtained for further research, including Metarhizium species, Coprinus cinerea, Trichoderma reesei, Schizophyllum commune, Laccaria bicolor, Tuber melanosporum, Postis placenta. By mapping the sequenced transcriptome reads to a reference genome, the structure annotation can be improved and novel transcripts and alternative splicing can be predicted, which expand the current genome databases. Proteomic one-dimensional or two-dimensional gel electrophoresis approaches combined with mass spectrometry have been widely used to identify interested proteins in fungi and many other organisms. Since the expression of some genes is regulated at the transcriptional level while some other genes at the translational level, the combination of transcriptome and proteome analysis is required and has been widely applied. In this study, we conducted the transcriptome and proteome analysis of different developmental stages of C. militaris and fruiting body cultured on rice medium). About 68 million reads were obtained from each sample and were mapped to the C. militaris genome. We found 2712 differentially expressed genes between mycelium and fruiting body. Functional annotations were used to investigate their difference including the cordycepin metabolism difference. In addition, 359 and 214 non-redundant proteins were identified from mycelium and fruiting body respectively by nESI-LC-MS/ MS. Proteome analysis revealed the developmental-associated protein expression patterns including some noteworthy proteins. Our transcriptome and proteome analysis data enrich the current database of C. militaris and will facilitate the future developmental study.
To upregulate cell-cell adhesion or aggregation associate with Ecadherin bind to a counterreceptor DARC
Leave a reply